






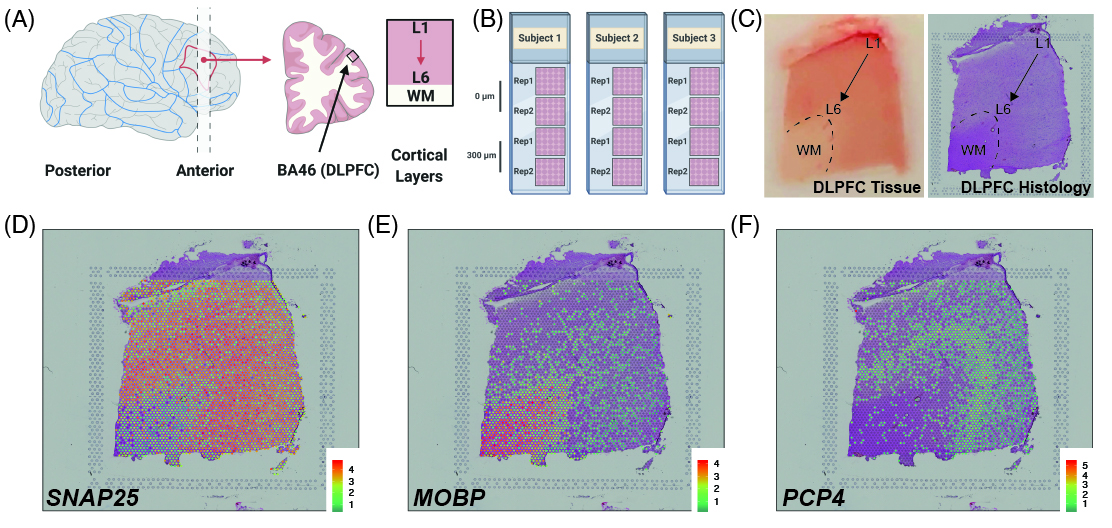
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
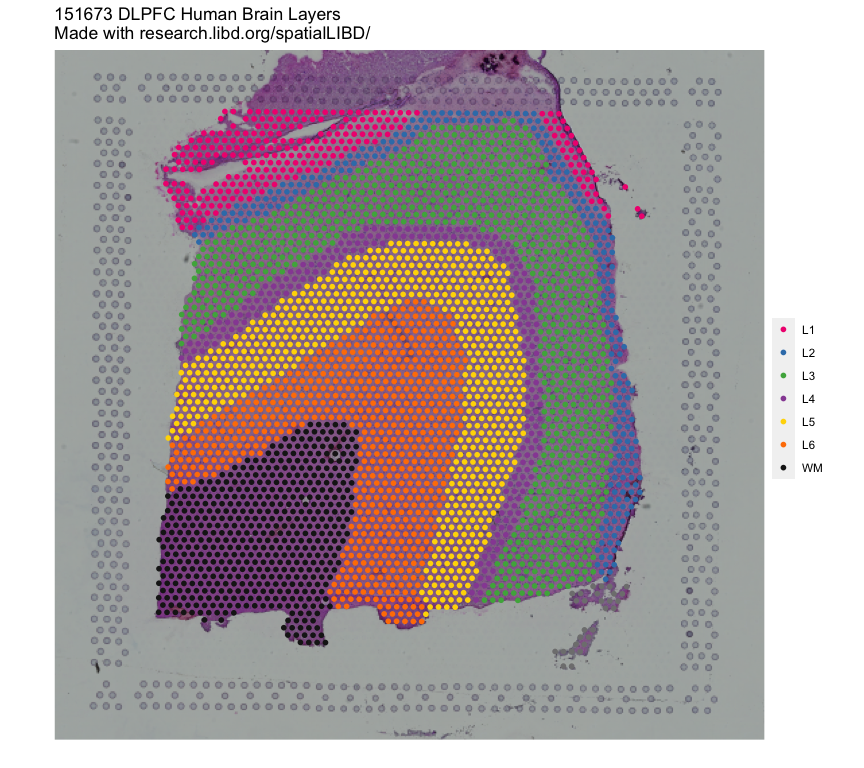
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Basic information overview about the spot-level SingleCellExperiment object. You can download it using
spatialLIBD::fetch_data(type = "spe").
When this information has been displayed it means that the shiny web application has finished loading and you can start exploring the rest of it.
Spot-level spatialLIBD documentation
This document describes the spot-level portion of the shiny web application made by the spatialLIBD Bioconductor package. You can either find the documentation about this package through Bioconductor or at the spatialLIBD documentation website. Below we explain the options common across tabs and each of the tabs at the spot-level data.
Slides and videos
You might find the following slides useful for understanding the features from this part of the web application.
These slides were part of our 2021-04-27 webinar for BioTuring that you can watch from their website or YouTube:
A recording of an earlier version of this talk is also available on YouTube.
You might also be interested in this video demonstration of spatialLIBD for the LIBD rstats club.
Raw summary
Before the documentation, this tab displays the SpatialExperiment object that contains the spot-level data. It’s basically useful to know that the data has been loaded and that you can start navigating the app. If you wish to download this data, use the following command.
## Download spe data
spe <- spatialLIBD::fetch_data(type = 'spe')
Throughout the rest of this document, we’ll refer to this object by the name spe.
Common options
Samples to plot: which sample to plot on the tabs that do not have grid on their name.Discrete variable to plot: which discrete variable (typically with the cluster labels) to visualize. We include the clusters:- the official manual annotation used extensively in our pilot study (DOI: 10.1038/s41593-020-00787-0)
- from the graph based clustering results produced by
spacerangerone Visium slide at a time. These clusters are saved asGraphBased. - from your own manual annotation of the spots under
ManualAnnotation. - resulting the manual annotation by Kristen R Maynard and Keri Martinowich using known markers and histology features. This was an initial exploration and was not analyzed in more detail.
- resulting from using a shared nearest neighbors approach with 50 neighbors cut at 4 up to 28 clusters. These are
SNN_k50_k4up toSNN_k50_k28. - described in Figure 7 from our paper (DOI: 10.1038/s41593-020-00787-0) such as
SpatialDE_PCA,SpatialDE_pool_PCAand others.
Reduced dimensions: which reduced dimension to visualize on theclusters (interactive)tab. Only the first two dimensions will be shown.Continuous variable to plot: which gene or continuous variable (such as the cell count, the ratio of the mitochondrial chromosome expression) to visualize in the gene tabs as well as on theclusters (interactive)tab.Gene scale: whether to use the raw expression values (counts) or the scaled and log transformed values (logcounts).Image name: the name of the background image to use. You can edit this image on theEdit imagetab.Spot transparency level: the transparency of the spots in the visualizations. It can be useful if the spot colors are blocking the background image.Minimum count value: Values from the selectedcontinuous variable to plotat or below this threshold will not be displayed.Gene color scale: Whether to use the color blind friendly palette (viridis) or to use a custom palette that we used for ourpaper. Other options from the viridisLite R package are also supported.Gene color direction: whether colors should be ordered from darkest to lightest or in the reverse direction.
We will cover the download button and upload CSV options at the end of this document.
Clusters (static)
Displays the selected cluster variable (from discrete variable to plot) for the given sample (from samples to plot). You can also choose to see the background image and clusters side to side.
## Reproduce locally with
spatialLIBD::vis_clus()
Clusters (interactive)
Displays a 1,200 by 1,200 pixels interactive plot area with a matrix of 2 by 2 plots. The top row shows the data at the spot-level with the histology information in the background. The bottom row shows the spot-level data at a reduced dimension space (PCA, TSNE, UMAP). The left column shows the selected gene or continuous variable, while the right column shows the selected cluster or discrete variable. The four plots are linked to each other such that if you use the lasso selector (mouse over to the top right of the interactive area to select it) in a single plot, the other 3 will get updated to highlight the same selection of points.
This panel allows you to look at the results from a given clustering approach and combine that information with the expression of a given gene to visualize the spot-level data both in the spatial resolution as well as a reduced dimensionality space from the expression of the most variable genes.
Once you have selected spots of interest, at the bottom of the tab there is a text box where you can enter your manual annotations. This overwrites spe$ManualAnnotation which is why you need to confirm doing so by clicking the button Label selected points (from lasso) with manual annotation. You can then change the clusters to plot option to ManualAnnotation to see your new spot labels.
Given the amount of data being displayed, this tab consumes quite a bit of resources.
Note that there can be a maximum of 36 unique manual annotations (unique values in spe$ManualAnnotation) before we run out of colors to visualize them.
Clusters grid (static)
This tab is similar to clusters (static) with the difference being that you can visualize a subset or even all samples at once. Select the samples you want to visualize, then specify the grid on which they will be plotted (the number of rows and columns), and click the upgrade grid plot button. We ask that you use this button every time you want to update the plot as it can take a few seconds for this update to complete.
This particular tab is useful if you have computationally defined some clusters using the data of all your samples and you want to visualize the resulting clusters across multiple (or even all the) samples at the same time.
## Reproduce locally with
spatialLIBD::vis_grid_clus()
Gene (static)
This tab is similar to clusters (static) but instead of displaying discrete values (like clusters), it displays continuous values such as the gene expression of a given gene or the number of cells per spot. By default, spots whose value is below or at 0 are not shown, which makes it easier for you to distinguish points with low values from those below the threshold of your interest (controlled by minimum count value). The points can be colored in two different color scales. You can also choose to see the background image and continuous values side to side.
## Reproduce locally with
spatialLIBD::vis_gene()
Gene (interactive)
This tab shows a single interactive plot. It is similar to clusters (interactive) as in you can use the lasso selector (mouse over top right to find it) to select spots and them label them using the text box at the bottom with the corresponding button. Unlike clusters (interactive), this version includes checkboxes at the top for each of the unique values of the selected discrete variable to plot such that you can subset the spots to those present on a given cluster.
Note that if you have discrete variable to plot toggled to the ManualAnnotation option and update the spot-level information with the text box and button at the bottom of the tab, then it will re-load the interactive visualization and you will lose your selection of points.
Gene grid (static)
This is the equivalent of clusters grid (static) but for continuous variables, just like gene (static) was the equivalent of clusters (static).
## Reproduce locally with
spatialLIBD::vis_grid_gene()
Edit image
This panel shows you all the options we have for manipulating the colors and properties of the selected background image. These results will be available under the edited_image option in the image chooser menu. You can perform sequential manipulations of the image, though it will get hard to keep track of all the changes you made.
## Reproduce locally with
spatialLIBD::img_update_all()
Saving and uploading your spe$ManualAnnotation results
Beyond visualizing the data, the main goal of this section of spatialLIBD is to enable you to label spots. That is done only through the interactive tabs as previously described. Your manual annotations are saved under spe$ManualAnnotation and you can save them for future use using the main menu download button. If you click this button, it will prompt you to save a CSV file. We recommend that you keep selected the checkbox Drop NA layer entries in the CSV file? which means that your CSV file will be smaller and will potentially lead to less conflicts with other CSV files you make. That is, you will likely avoid re-labeling the same spot with different values in two or more of these CSV files. This is particularly useful if you plan to work on one sample at a time, save your results, and then merge them all into a single CSV file.
These CSV files with your manual annotations can be re-uploaded to spatialLIBD. You can notice this by choosing ManualAnnotation under clusters to plot and using any of the clusters tabs. If you upload more than one CSV, any values you have under spe$ManualAnnotation will be overwritten if the spot is present in your CSV file. Thus, if you followed the recommended workflow of saving one CSV file per sample, you can then upload them all sequentially and merge them together into a single CSV file to simplify your work later.
In summary, the order in which you re-upload the CSV files matters as newer uploads will overwrite any duplicated spots from previous CSV files.
We also recommend saving your work often in case you lose connection to spatialLIBD. Though you could always run this website locally by using the following command:
## Reproduce locally with
spatialLIBD::run_app()
This will require about 3GB of RAM to run on the server side, though potentially more, specially when using the clusters (interactive) tab.
Download PDF
Click the 'upgrade custom image' button above to save the custom image. You can then select 'edited_image' and use it in other parts of the web application. Note that if you had 'edited_image' already selected, you'll need to re-select or change another input to update the other plots.